Solid Biosciences annuncia l’autorizzazione FDA IND per la prima terapia genica a doppia via di somministrazione nel settore per il trattamento delle manifestazioni neurologiche e cardiache dell’atassia di Friedreich
CHARLESTOWN, Massachusetts., 07 gennaio 2025(GLOBE NEWSWIRE)
Società Solid Biosciences Inc.(Nasdaq: SLDB) (la “Società” o “Solid”), una società di scienze della vita che sviluppa medicinali genetici di precisione per malattie neuromuscolari e cardiache, ha annunciato oggi che la Amministrazione per gli alimenti e i farmaci degli Stati Uniti(FDA) ha autorizzato la sua domanda di farmaco sperimentale (IND) per SGT-212 per il trattamento dell’atassia di Friedreich (FA), una malattia degenerativa causata da livelli insufficienti della proteina fratassina. SGT-212 è il nuovo candidato alla terapia genica FA basata su AAV della Società, progettato per fornire fratassina a lunghezza intera tramite infusione endovenosa sistemica (IV) e infusione diretta di nuclei intradentati (IDN) nel cervelletto. SGT-212 è progettato per trattare le manifestazioni cliniche neurologiche e sistemiche di FA per affrontare l’intero spettro di progressione della malattia.
La FA è una malattia multisistemica altamente complessa che presenta sfide distinte per lo sviluppo di farmaci, in parte perché la fratassina è una proteina che richiede: (1) livelli di espressione precisi per evitare tossicità cardiache fatali e (2) localizzazione tissutale sul bersaglio nel cervelletto per ottenere potenziali benefici clinici neurologici. SGT-212 è l’unico candidato in fase di sviluppo che utilizza due vie di somministrazione per affrontare le manifestazioni cardiache della FA, erogando anche direttamente la terapia ai nuclei dentati nel cervelletto, la regione più colpita e implicata nel declino neurologico associato alla FA.
Bo Cumbo, Presidente e CEO di Bioscienze solide, ha commentato: “SGT-212 è stato intenzionalmente progettato per consentire una somministrazione altamente mirata della nostra terapia genica sia ai nuclei dentati che al tessuto cardiaco. L’IND è stato supportato da un solido pacchetto preclinico che dimostra una trasduzione sicura e l’espressione della fratassina in questi tessuti bersaglio, con un significativo ripristino della funzione neurologica e l’inversione delle implicazioni cardiache del disturbo nei topi. Nel corso degli anni, abbiamo testato diversi candidati utilizzando diversi metodi di somministrazione e abbiamo condotto molteplici studi NHP, alcuni dei quali si sono estesi per un anno. Sulla base di questa ricerca, crediamo che una doppia via di somministrazione mirata a più sistemi sia il miglior approccio in fase di sviluppo per affrontare direttamente le implicazioni neurologiche che hanno un impatto profondo sulla vita quotidiana dei pazienti, mirando contemporaneamente alle manifestazioni cardiache che svolgono un ruolo chiave nella malattia più avanzata. SGT-212 offre un approccio veramente differenziato per affrontare l’AF con il potenziale di trattare l’intero spettro di sintomi e speriamo di incontrare ogni paziente nel punto in cui si trova nel corso della sua malattia FA”
Farmer Jennifer, Amministratore Delegato di Friedreich’s Alleanza per la ricerca sull’atassia(FARA), ha aggiunto: “Ci congratuliamo Bioscienze solide al raggiungimento di questo importante traguardo. Gli approcci alla terapia genica sono mirati alle cause sottostanti della FA e quindi importanti nella strategia complessiva per trattare e curare questa malattia. Ci sono stati progressi incoraggianti nel panorama del trattamento della FA; tuttavia, c’è ancora un bisogno medico insoddisfatto per la nostra comunità di pazienti. Attraverso il nostro lavoro con le persone che vivono con la FA e le loro famiglie, sappiamo che cercano terapie progettate per trattare i sintomi neurologici debilitanti che le persone che vivono con la FA affrontano quotidianamente, come la perdita di deambulazione e coordinazione, la disartria, insieme alla malattia cardiaca che accorcia la vita. L’approccio unico e preciso di SGT-212 prende di mira sia il cervelletto che il tessuto cardiaco utilizzando una doppia via di somministrazione e, così facendo, mira ad affrontare la causa sottostante della malattia e la progressione della FA. Non vediamo l’ora di continuare la partnership con l’azienda mentre avanzano SGT-212 nella clinica più avanti quest’anno.”
Nella seconda metà del 2025, la Società prevede di avviare uno studio clinico di Fase 1b first-in-human, in aperto, di determinazione della dose di SGT-212. Lo studio arruolerà pazienti adulti non deambulanti e deambulanti affetti da FA in un massimo di tre coorti e valuterà la sicurezza e la tollerabilità della somministrazione contemporanea di IDN sistemica e bilaterale di SGT-212. I partecipanti allo studio saranno seguiti fino a cinque anni dopo aver ricevuto SGT-212.
Informazioni su SGT-212
SGT-212 è una terapia di sostituzione genica basata su AAV ricombinante per l’atassia di Friedreich (FA) progettata per fornire fratassina umana (Fxn) a lunghezza intera tramite una doppia via di somministrazione: infusione nel nucleo intradentato (IDN), utilizzando un dispositivo guidato da MRI, seguita da un’infusione endovenosa (IV) per aumentare i livelli terapeutici di Fxn rispettivamente nei nuclei dentati cerebellari e nei cardiomiociti. Si prevede che il ripristino dei livelli di Fxn ripari la disfunzione mitocondriale sottostante nei neuroni e nei cardiomiociti per affrontare sia le manifestazioni neurologiche che cardiache della malattia.
SGT-212 è stato sviluppato da Società a responsabilità limitata FA212 LLC, un’azienda fondata da genitori di bambini affetti da FA,Università della Pennsylvania, EBio scienze solide.
Divulgazione finanziaria dell'Università della Pennsylvania
Il laboratorio di Dottor Wilsonal Università della Pennsylvania ha ricevuto finanziamenti per la ricerca sponsorizzata da Società a responsabilità limitata FA212 LLC. Penn possiede una quota azionaria in Società a responsabilità limitata FA212 LLC Penna e Dottor Wilson hanno ricevuto, o potrebbero ricevere in futuro, una contropartita finanziaria relativa alla concessione in licenza di determinate proprietà intellettuali della Penn a Bioscienze solide.
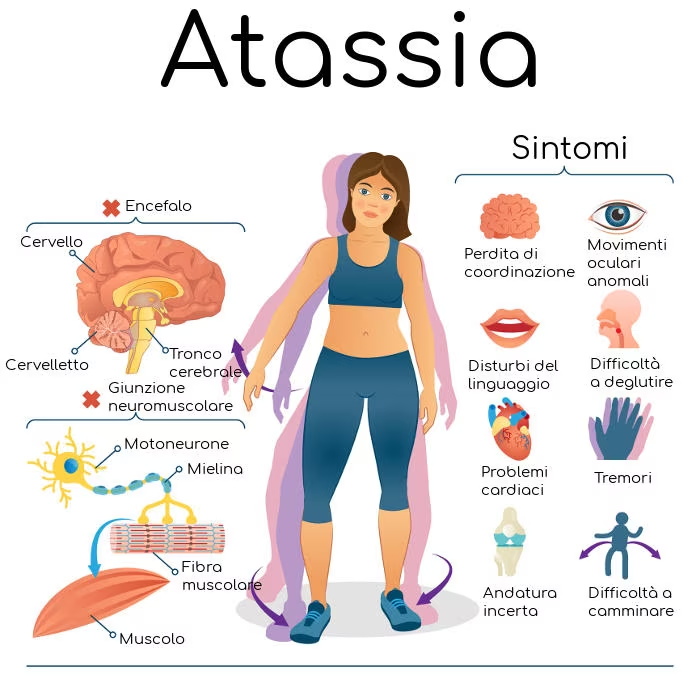
Informazioni sull'atassia di Friedreich (FA)
La FA è una malattia multisistemica degenerativa ereditaria e potenzialmente letale causata da difetti nel gene della fratassina che interrompono la produzione della proteina fratassina, una proteina mitocondriale legante il ferro coinvolta in processi cellulari essenziali, tra cui la produzione di energia. È noto che la FA causa danni progressivi al sistema nervoso, problemi di movimento e disfunzione cardiaca, con complicazioni cardiache identificate come causa primaria di morte. La FA colpisce circa 5.000 persone in gli Stati Uniti e 15.000 in Europa Attualmente non esistono trattamenti che possano curare o arrestare la progressione della malattia.
Informazioni su FARA
I Friedreich Alleanza per la ricerca sull’atassia(FARA) è un’organizzazione nazionale senza scopo di lucro dedicata alla cura dell’atassia di Friedreich (FA) attraverso la ricerca. Le sovvenzioni e le attività FARA forniscono supporto per la ricerca di base e traslazionale sull’atassia, lo sviluppo di farmaci farmaceutici/biotecnologici, le sperimentazioni cliniche e le conferenze scientifiche. FARA funge anche da catalizzatore, tra il pubblico e la comunità scientifica, per creare scambi di informazioni in tutto il mondo che guidino i progressi medici. Per maggiori informazioni su FARA, visita www.curefa.org .
Di Bioscienze solide
Bioscienze solide è un’azienda di medicina genetica di precisione focalizzata sullo sviluppo di un portafoglio di candidati alla terapia genica, tra cui SGT-003 per il trattamento della distrofia muscolare di Duchenne (Duchenne), SGT-212 per il trattamento dell’atassia di Friedreich, SGT-501 per il trattamento della tachicardia ventricolare polimorfica catecolaminergica (CPVT), SGT-601 per il trattamento della cardiomiopatia dilatativa mediata da TNNT2, SGT-401 per il trattamento della cardiomiopatia dilatativa mediata da BAG3 e risorse aggiuntive per il trattamento di malattie cardiache fatali. Solid sta sviluppando la sua pipeline diversificata in malattie neuromuscolari e cardiache rare, riunendo esperti in scienza, tecnologia, gestione delle malattie e assistenza. Incentrata sul paziente e fondata da coloro che sono direttamente colpiti, il mandato di Solid è quello di migliorare la vita quotidiana dei pazienti che convivono con queste malattie devastanti. Per maggiori informazioni, visita www.solidbio.com .
Dichiarazioni previsionali
Il presente comunicato stampa contiene “dichiarazioni previsionali” ai sensi del Private Securities Litigation Reform Act del 1995, tra cui dichiarazioni relative alle aspettative, ai piani e alle prospettive future per la Società; la capacità di raggiungere e attuare con successo gli obiettivi e le priorità della Società e di raggiungere traguardi clinici chiave; la pipeline di programmi della Società per le malattie neuromuscolari e cardiache, inclusi i suoi programmi SGT-212 e SGT-003 e le aspettative per le richieste CTA, le attivazioni dei siti, lo sviluppo clinico, l’inizio e l’arruolamento negli studi clinici, il dosaggio, la disponibilità dei dati degli studi clinici e la potenziale approvazione accelerata; la sufficienza della liquidità, delle disponibilità liquide e dei titoli disponibili per la vendita della Società per finanziare le sue operazioni; e altre dichiarazioni contenenti le parole “anticipare”, “credere”, “continuare”, “potrebbe”, “stimare”, “aspettarsi”, “intendere”, “potrebbe”, “pianificare”, “potenziale”, “prevedere”, “progettare”, “dovrebbe”, “obiettivo”, “vorrebbe”, “lavorare” ed espressioni simili. Tutte le dichiarazioni previsionali si basano sulle aspettative attuali della direzione su eventi futuri e sono soggette a una serie di rischi e incertezze che potrebbero causare risultati effettivi sostanzialmente diversi e sfavorevoli da quelli stabiliti o impliciti in tali dichiarazioni previsionali. Questi rischi e incertezze includono, ma non sono limitati a, rischi associati alla capacità dell’azienda di far avanzare SGT-212, SGT-003, SGT-501, SGT-601, SGT-401 e altri programmi preclinici e librerie di capsidi nei tempi previsti o in assoluto; ottenere e mantenere le necessarie approvazioni dalla FDA e da altre autorità di regolamentazione; replicare negli studi clinici i risultati positivi riscontrati negli studi preclinici e negli studi clinici in fase iniziale dei prodotti candidati dell’azienda; ottenere, mantenere o proteggere i diritti di proprietà intellettuale relativi ai suoi prodotti candidati; competere con successo con altre aziende che cercano di sviluppare la distrofia di Duchenne, l’atassia di Friedreich e altri trattamenti neuromuscolari e cardiaci e terapie geniche; gestire le spese; e raccogliere il sostanziale capitale aggiuntivo necessario, nei tempi necessari, per continuare lo sviluppo di SGT-212, SGT-003, SGT-501, SGT-601, SGT-401 e altri candidati, raggiungere i suoi altri obiettivi aziendali e continuare come azienda in funzionamento. Per una discussione di altri rischi e incertezze e altri fattori importanti, ognuno dei quali potrebbe causare risultati effettivi dell’azienda diversi da quelli contenuti nelle dichiarazioni previsionali, vedere la sezione “Fattori di rischio”, nonché discussioni di potenziali rischi, incertezze e altri fattori importanti, nei documenti più recenti dell’azienda presso la Commissione per i titoli e gli scambi. Inoltre, le dichiarazioni previsionali incluse nel presente comunicato stampa rappresentano le opinioni della società alla data odierna e non devono essere considerate come rappresentative delle opinioni della società in qualsiasi data successiva alla data odierna. La società prevede che eventi e sviluppi successivi causeranno un cambiamento delle opinioni della società. Tuttavia, mentre la società può scegliere di aggiornare queste dichiarazioni previsionali in un momento futuro, la società declina espressamente qualsiasi obbligo di farlo.
Contatto per gli investitori di Solid Biosciences:
Nicole Anderson
Direttore, Relazioni con gli investitori e Comunicazioni aziendali
Società Solid Biosciences Inc.
investitori@solidbio.com
Contatto con i media:
Glenn Argento
Partner FINN
glenn.silver@finnpartners.com